
SARS 病毒测序
2003 年 4 月,我们在基因组科学中心 (GSC) 公开发布了首个完整的冠状病毒序列组装结果,该病毒现在被认为是导致严重急性呼吸综合征 (SARS) 的原因。自 1999 年成立以来,GSC 一直使用 Linux 来进行所有分析、存储和网络基础设施。SARS 项目的序列数据存储、处理和公开分发均通过多台 Linux 服务器完成,从功能强大且轻薄的 IBM x330 到巨型的八路 Xeon x440。Linux 提供了灵活的基础设施,使我们能够自动化测序流程中的几乎每个过程。在 Linux 社区通过新闻组、Web 文章和 HOWTO 的支持下,我们能够以极其经济高效的方式利用商品化和中档硬件。
自 2002 年 11 月 16 日首次记录 SARS 病例以来,该病毒已在中国(92%)、加拿大(3%)、新加坡(2%)和美国(1%)以及其他 25 多个国家/地区造成共 8,458 例病例。SARS 的死亡率约为 5-10%,60 岁以上人群的死亡率高达 50%。截至 2003 年 6 月 24 日,SARS 已夺走 807 条生命,并对受影响地区的经济产生了深刻的负面影响——仅中国就可能因旅游业和税收损失数十亿美元的收入。
2003 年 3 月 27 日,我们中心的主任 Marco Marra 和我们的项目负责人 Caroline Astell 决定对 SARS 冠状病毒进行测序。2003 年 4 月 7 日凌晨 1 点,大约 50ng 来自多伦多患者的病原体 Tor2 分离株的遗传物质从加拿大温尼伯的 4 级国家微生物实验室运抵。五天后,即 2003 年 4 月 12 日,我们在 Apache 服务器上的 Zope/Plone 页面上发布了 Tor2 分离株 (Tor2/SARS) 冠状病毒序列的 29,751 个碱基组装结果,供公众访问。几天后,亚特兰大佐治亚州 (疾病控制中心) CDC 发布了 Urbani 分离株的序列。
在 1990 年代之前,快速收集大量序列信息的技术尚不存在。人类基因组计划 (HGP) 于 1991 年启动,到 1999 年仅收集了 15% 的序列。然而,得益于 1990 年代开发的新方法,HGP 急剧转向完成。到 2000 年年中,90% 的人类序列已可用,目前基因组序列基本上已完成。来自 HGP 等测序项目的数据存储在 NCBI 的 Genbank 中并可公开访问。

图 1. 我们的测序实验室全景图:1) 代表流程的条形码,2) Tango 液体处理平台,3) –112°F 冷冻机,4) 热循环仪电源,5) ABI 3730XL 测序仪,6) ABI 3700 测序仪,7) x330 测序仪控制集群,8) 网络/电源连接和 9) 测序仪通风管道。
在其运营的头十年(1982-1992 年)中,Genbank 仅收集了 80,000 条记录中略高于 100MB 的序列。在接下来的十年(1992-2002 年)中,Genbank 的增长率飞速上升,数据库增长到 29GB——是人类基因组大小的十倍——共 2200 万条记录。Genbank 每天从世界各地的测序实验室接收约 10,000 条序列记录。其中一个实验室是 GSC,它于 2003 年 4 月 13 日将 Tor2/SARS 的序列存入 Genbank。要了解 Linux 如何参与导致序列 gi:29826276 提交的过程,我们需要回到起点。
1999 年 6 月,该实验室由六台米色机箱计算机和人数相同的员工组成。中央文件服务器(2xP3-400、512MB RAM、Red Hat 5.2 和 2.0.36 内核)使用 DPT IV 卡提供三个 RAID-0 18GB SCSI 磁盘。第二台机器 (P3-400) 导出了另外 50GB 的软件 RAID。凭借其他三台 Linux 客户端和一台 Microsoft Windows NT 工作站,这些机器位于 BC 癌症机构 (BCCA) 网络上。

图 2. 第一代服务器硬件:1) VA Linux VAR900 2xXeon-500 导出 1TB,2) Raidion.u2w RAID 控制器,3) 2x8x36GB SCSI 磁盘和 4) VA Linux 2230s 和 3x10x72GB SCSI 磁盘。
我们起步的时机对我们有利。像所有研究实验室一样,我们需要共享磁盘、分配进程、编译软件以及存储和整理数据。换句话说,所有 UNIX 擅长的事情。如果我们早 2-3 年开始,采用新兴的 Linux 将会很困难。很可能,我们现在不是将廉价的旧 PC 降级为办公室或不太密集的网络任务,而是会尝试最大化我们对老化的 Sun 服务器的大量投资回报。幸运的是,结果证明可以购买相对便宜的 PC,安装 Linux 并拥有一个强大、灵活且极其经济高效的 UNIX 环境。多亏了 Linux,不再需要花费整个薪水购买 UNIX 工作站。
现在是选择 Linux 的好时机。2.0 内核非常稳定;NFS 服务器正在稳定,并且可以选择功能齐全的桌面环境。我们能够下载或编译生物信息学分析的基本工具箱,例如 HGP 的开源主力:BLAST(序列比较)、Phred(测序仪产生的轨迹的碱基识别)、Phrap(序列组装)和 Consed(序列组装可视化),以及各种序列和蛋白质数据库。当然,Perl 填补了任何空白。我们完成计算工作的入门成本很低,我们可以更有效地花费拨款资金来扩展实验室(图 1)。
1999 年秋季,我们收到了第一台 DNA 测序仪 MegaBACE 1000(图 6)。测序仪确定 DNA 样本的特定碱基序列,尽管目前技术仅限于一次准确确定 500-800 个碱基。这个读取长度远小于最小的基因组的大小(Tor2/SARS 的大小为 30,000 个碱基)。因此,测序仪一次同时处理 96 个样品,有些可以装载多个 96 孔或 384 孔板。
MegaBACE 是一种 SCSI 设备,Applied Biosystems (ABI) 3700 和 3730XL 测序仪(图 6)通过串行接口控制,并通过以太网连接发送数据。虽然这些测序仪以自动化方式获取大量数据,但它们的软件是一个点击式 Windows 应用程序。基于 UNIX 的控制应用程序将彻底改变这些机器的部署,尤其是在大型实验室中。我们已经通过部署 IBM x330 来替换测序仪随附的原始 PC,从而降低了 3700 的维护复杂性(图 6)。将 Windows 测序平台集成到 Linux 网络中是 smbmount、rsync、Perl 和 Apache 的完美工作。在每次测序运行结束时,操作员会触发一个 Web 控制的数据镜像过程,将任何新数据复制到网络磁盘上。
镜像完成后,文件首先从其专有格式(编码原始信号轨迹)转换为实际碱基及其相关质量度量,然后存储在 MySQL 数据库 (3.23.55max) 中。到目前为止,我们已经收集了大约 200 万个测序读数,或大约 1TB 的原始序列数据。
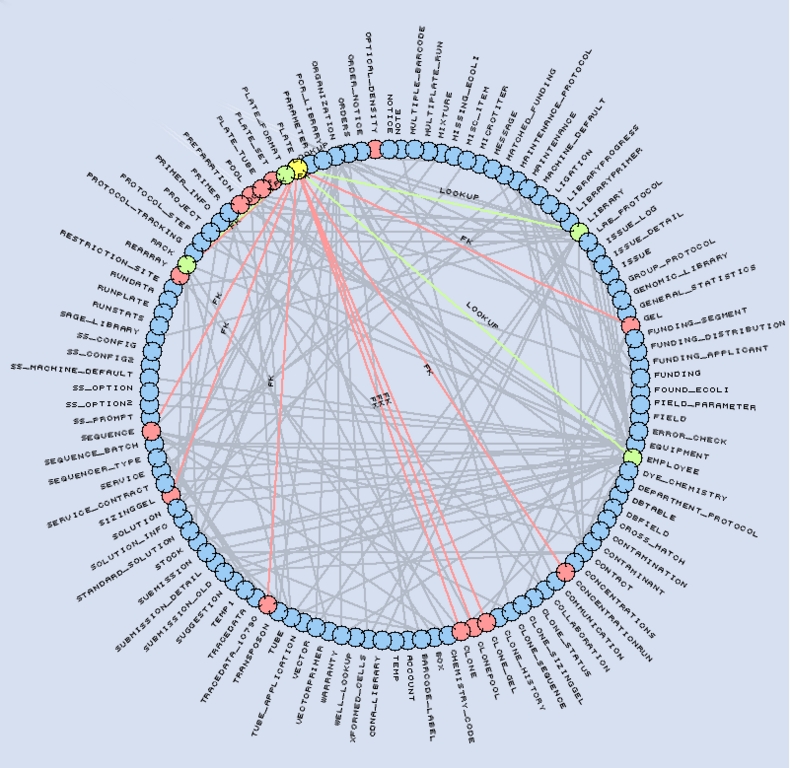
图 3. 我们的 LIMS 架构表示。Plate 表(黄色)引用了四个表(绿色),并被 14 个表(红色)引用。
MySQL 实验室信息管理系统 (LIMS) 数据库是我们测序过程的核心。它的架构包含 115 个表、1,171 个字段和 195 个外键。该数据库跟踪实验室中执行的所有试剂、设备、流程和反应。我们通过使用应用程序逻辑和特定的字段命名约定来规避 MySQL 缺少本机外键支持的问题。外键命名为 FKTYPE_TABLE__FIELD,表示它们指向 TABLE 表中的 TABLE_FIELD。外键名称的可选 TYPE 部分用于支持指向同一 TABLE_FIELD 的多个键。
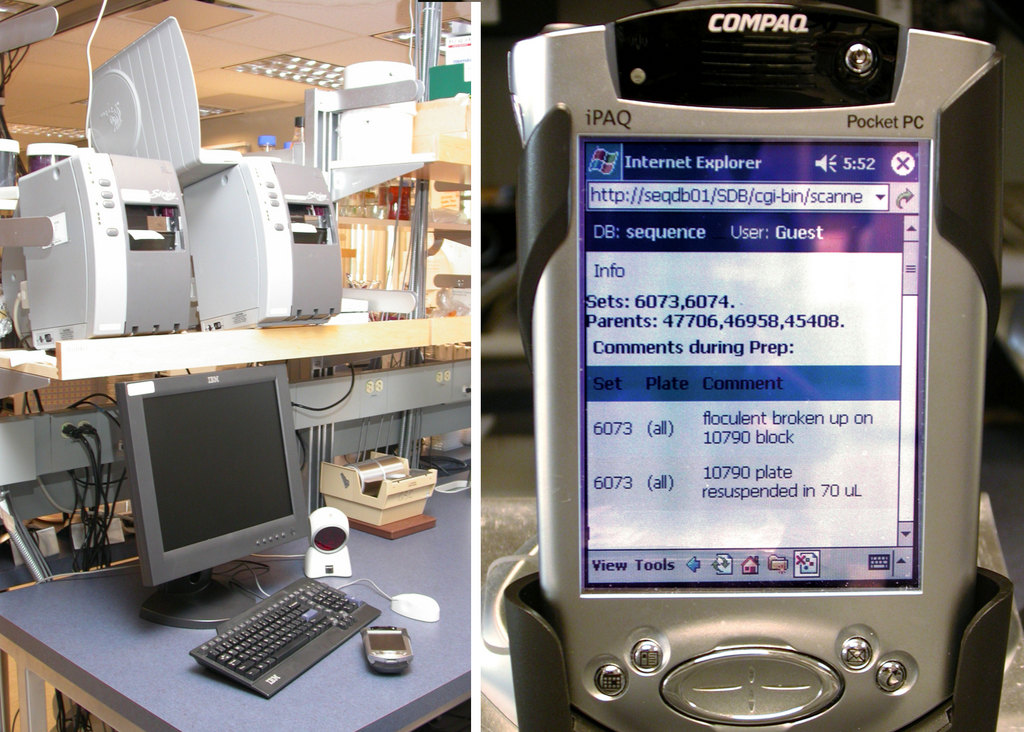
图 4. 条形码使用联网的 Zebra 打印机打印(左图)。iPAQ 提供到 LIMS 的移动界面(右图)。

图 5. 实验室中几乎所有东西都贴有条形码。
实验室技术人员使用配备条形码扫描仪的 Wi-Fi Compaq iPAQ 与 LIMS 数据库进行交互(图 4)。iPAQ 连接到我们的内部 Apache Web 服务器,该服务器为一套 mod_perl 脚本提供支持。解决方案、板和设备等对象都贴有条形码(图 5)。条形码打印在联网的 Zebra S600/96XiIII 条形码打印机(图 4)上,这些打印机使用高粘性标签进纸,这些标签在我们的 –112°F 冷冻机中保持粘附性。条形码软件是用 Perl 编写的,使用 ZPL 打印机语言来格式化标签,并使用 lpr 分发打印。

图 6. 测序仪:1) MegaBACE 1000,2) 测序仪 PC,3) UPS,4) 测序仪电源,5) ABI 3700s,6) ABI 3730XL 和 7) x330 测序仪集群。
自 MegaBACE 1000 以来,三代测序仪已经过我们的实验室,我们目前运行六台 ABI 3700 和三台 ABI 3730XL(图 6)。最新的 ABI 3730XL 能够接受多个 384 孔板,并在 24 小时内测序 1,152 个 DNA 样品。每个样品产生多达 700-800 个高质量碱基,一台 3730XL 每天产生约 800,000 个碱基。
Tor2/SARS 基因组是使用全基因组鸟枪法 (WGS) 测序的。在这种方法中,基因组的随机片段以冗余方式测序,然后组装在一起以恢复整个基因组序列。鉴于病原体的大小预计约为 30,000 个碱基,因此至少需要 40 个读数才能跨越基因组。然而,由于读数源自随机区域,因此需要比完整组装所需的最小读数数量更多。冗余还提高了确定基因组中每个给定位置碱基的置信度。
当我们购买第一批 IBM x330 服务器时,现在是 168-CPU 集群的一部分(图 7),1U 平台正处于进入商业现货 (COTS) 类别并开始享受 COTS 价格的边缘。米色机箱不再用于分布式计算。负载重的生产子系统(如 Apache 和 MySQL)安装在 IBM 的 4U x440 上,后者是具有 8GB RAM 的八路超线程 Xeon 节点。这些机箱运行 SuSE 8.1——少数几个支持 IBM Summit 芯片组的发行版之一。x440 是一台 NUMA 机器,每个四 CPU 模块具有 32MB 的 L4 缓存,如果没有 IBM 的 Summit 补丁,内核只能识别出两个 CPU。SuSE 基于 2.4.19 的派生内核,具有 bigmem+Summit 支持,可以利用所有八个 CPU 和 8GB 内存。即使没有 2.5 系列内核中现在的高级 NUMA 调度程序代码,这些 x440 也一直是真正的主力,使我们能够并行运行八个 BLAST 进程,并有足够的 RAM 将整个人类基因组缓存在共享内存中。任何声称 Linux 尚未为大型机做好准备的人都会感到惊讶。

图 7. 计算和存储基础设施:1) 2001 年 1 月 x330 的初始部署,2) 84 台 x330 节点和 3) NetApp FAS960 文件管理器和两个 IBM 3583 LTO 库,每个库有两个驱动器和 72 个插槽,由在 x342 上运行的 Veritas 控制。
随着我们的快速增长,NFS 子系统开始出现问题。特别是,机器在某些 NFS 服务器/客户端版本组合中崩溃。虽然根据我们的经验,NFS 客户端是稳健的,但当前的 Linux NFS 服务仍有改进空间。我们最快的 NFS 服务器 IBM x342 (2xP3-1.24, 2GB RAM) 无法处理超过 4,000-6,000 次 NFS 操作/秒,尤其是在来自我们集群的大量并行读/写期间。为了解决性能限制,我们购买了 NetApp FAS960 Filer(图 7)。该文件管理器具有 10TB 的原始存储空间 (5x14x144GB),已达到 30,000 次 NFS 操作/秒。尽管存在 NFS 问题,但我们最初的 VAR900 生产文件服务器(图 2)是稳定性的典范,在 2002 年 2 月重启进行升级之前,其正常运行时间达到了 394 天。
第一批 Tor2/SARS 序列数据于 2003 年 4 月 11 日星期五晚上可供我们的信息学小组分析。为了验证我们的序列反应,我们检查了它是否受到污染。BLAST 搜索使我们能够确定公共蛋白质组和基因组数据库中最接近的匹配项。令我们欣慰的是,最佳匹配项是牛冠状病毒(图 8),这表明我们正在测序与冠状病毒相关的物质。这些病毒的序列以一串 A 结尾,当我们看到以 poly-A 尾结尾的序列读数时,我们确信这是基因组的一端。

图 8. 来自 BLAST 查询的 Top Hit 输出
x330 和 x440 用于分析和组装 SARS 数据。基因组不是很大,组装在单个 CPU 上花费不到 15 分钟。相比之下,第一个公开组装的人类基因组,是 Tor2/SARS 大小的 300,000 倍,是在 UCSC 完成的,在一个 100-CPU Linux 集群上花费了四天时间。
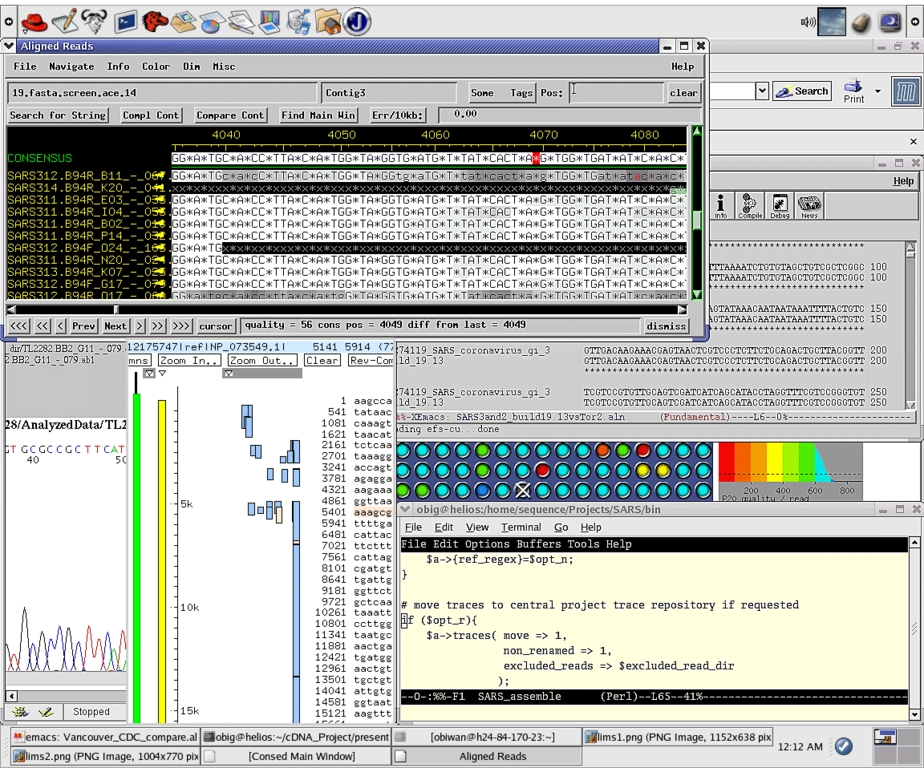
图 9. Linux 桌面上的序列分析

图 10. 其中一个 SARS 板的序列读取质量
到 2003 年 4 月 12 日星期六凌晨 2:25,我们完成了 Tor2/SARS 的第七次构建,并将此组装冻结为第一个草案。它被导入 AceDB 以可视化与其他已知蛋白质集的比对以进行验证(图 9)。我们花费了星期六的时间来验证组装,该组装于当天晚些时候使用在 Zope/Plone 下运行的自定义 CMS 系统发布到我们的 x440 公共 Web 服务器上。
Tor2/SARS 的序列已经鉴定了第四个新的冠状病毒组,并提供了开发诊断测试以及可能的疗法(包括疫苗)的必要信息。Linux 使我们能够在不花费大量硬件或软件资金的情况下完成我们的工作。使用商品化硬件最大限度地减少了因实施时间长而造成的折旧损失。我们将关注要捕获的新错误,与此同时,我们的 MySQL 数据库已开放用于测序。
作者要感谢 Marco Marra、Steven Jones、Caroline Astell、Rob Holt、Angela Brooks-Wilson、Jas Khattra、Jennifer Asano、Sarah Barber、Susanna Chan、Allison Cloutier、Sean Coughlin、Doug Freeman、Noreen Girn、Obi Griffith、Steve Leach、Mike Mayo、Helen McDonald、Steven Montgomery、Pawan Pandoh、Anca Petrescu、Gord Robertson、Jacquie Schein、Asim Siddiqui、Duane Smailus、Jeff Stott 和 George Yang 在科学专业知识、实验室和生物信息学工作方面的贡献。我们还要感谢 Kirk Schoeffel、Mark Mayo 和 Bernard Li 在系统管理方面的建议。
命令行生物信息学
让我们使用 bash 和 /bin 和 /usr/bin 中的一些二进制文件进行一些生物信息学分析。我们将计算 Tor2/SARS 基因组的 GC 比率——即 G 或 C 碱基对的比例。让我们避免使用 awk 来使事情变得有趣。首先,使用 wget 下载序列,使用 -q 静音其详细输出
> wget -q http://mkweb.bcgsc.ca/sars/AY274119.fa > head AY274119.fa gi|30248028|gb|AY274119.3| SARS coronavirus TOR2 ATATTAGGTTTTTACCTACCCAGGA...
序列文件采用 FASTA 格式,由标题行和序列组成,序列拆分为固定宽度的行。以下代码计算序列中 G 和 C 的数量,并将总数表示为碱基总数的百分比
> grep -v "^>" AY274119.fa | fold -w 1 | tr "ATGC" "..xx" | sort | uniq -c | sed 's/[^0-9]//g' | t -s "\012" " " | sed 's/\([0-9]*\) \([0-9]*\)/scale = 3; ↪\2 \/ (\1+\2)/' | bc -i scale = 3; 12127 / (17624+12127) .407
在我们的序列中的 29,751 个碱基中,有 12,127 个是 G 或 C,GC 含量为 41%。
GSC MySQL LIMS
我们收集了 3,250 个测序读数,其中包含 210 万个高质量碱基对,为初始草案组装做出了贡献。这代表了大约 70 倍的基因组冗余覆盖率。WGS 通常最多完成 10 倍,但对我们来说,时间至关重要,我们希望避免与完成第一轮测序未完全覆盖的区域相关的延迟。
SELECT SUM(Sequence_Length) AS bp_tot, AVG(Quality_Length) AS bpq_avg, SUM(Quality_Length) AS bp_qual_tot, COUNT(Well) AS reads, Sequence_DateTime AS date, Equipment_Name AS equip FROM Equipment, Clone_Sequence, Sequence_Batch, Sequence, Plate, Library, Project WHERE FK_Sequence_Batch__ID=Sequence_Batch_ID AND FK_Plate__ID=Plate_ID AND FK_Library__Name=Library_Name AND FK_Equipment__ID=Equipment_ID AND FK_Project__ID=Project_ID AND FK_Sequence__ID=Sequence_ID AND Sequence_Subdirectory like "SARS2%" AND Quality_Length > 100 AND Sequence_DateTime < "20030413" GROUP BY Sequence_ID ORDER BY Sequence_DateTime; bp_tot bpq_avg bp_tot reads date equip 437256 612.6399 205847 336 2003-04-11 21:07:06 SARS212.B21 D3730-3 412366 752.1074 245187 326 2003-04-11 22:15:34 SARS213.B21 D3730-1 269456 639.1926 225635 353 2003-04-11 22:22:34 SARS215.B21 D3700-6 130525 715.5060 118774 166 2003-04-11 22:25:44 SARS216.B21 D3700-5 282490 682.6311 249843 366 2003-04-11 22:27:14 SARS215.BR D3700-4 310119 612.7601 212015 346 2003-04-11 22:31:56 SARS213.BR D3700-1 182573 681.4975 136981 201 2003-04-11 22:36:40 SARS216.BR D3700-3 301471 642.2273 226064 352 2003-04-12 01:58:16 SARS212.BR D3700-2 401595 690.5204 220276 319 2003-04-12 05:13:26 SARS211.BR D3730-3 460100 642.0468 219580 342 2003-04-12 06:20:52 SARS214.BR D3730-2 182360 471.7832 67465 143 2003-04-12 07:14:44 SARS214.B21 D3730-1
资源
Genbank 的增长: www.ncbi.nlm.nih.gov/Genbank/genbankstats.html
Perl 如何拯救人类基因组计划: bioperl.org/GetStarted/tpj_ls_bio.html
冠状病毒图像: www3.btwebworld.com/vdg/gallery/Coronavirus.jpg
Science 杂志的 SARS 特刊: www.sciencemag.org/feature/data/sars
SARS 统计数据和信息: www.cdc.gov/ncidod/sars 和 lassesen.com/sars
SARS 历史时间线: www.worldhistory.com/sars.htm
UCSC 人类基因组组装: www.cse.ucsc.edu/~learithe/browser/goldenPath/algo.html
Martin Krzywinski (martink@bcgsc.ca) 是加拿大 Michael Smith 基因组科学中心的一名生物信息学研究科学家。他将时间用于将 Perl 应用于物理图谱和数据处理自动化方面的问题。在他的空闲时间,他可以被发现鼓励他的猫坚持她的饮食。
Yaron Butterfield (ybutterf@bcgsc.ca) 领导加拿大 Michael Smith 基因组科学中心的测序生物信息学团队。他和他的团队为各种基因组和基于癌症的研究项目开发 DNA 序列分析和可视化软件及流程。

{kind=link}